
Nicht immer ist die Diagnose auch nach Einschätzung durch die Task-Force-Diagnosekriterien klar. Oft ist es nicht ganz einfach, ARVC von anderen Herzerkrankungen abzugrenzen. Nach den wichtigsten Differentialdiagnosen und Erkrankungen, mit denen Overlaps möglich sind, werden einige wichtige Differentialdiagnosen weiter unten näher erläutert.
- Myokarditis (Herzmuskelentzündung)
- Sarkoidose
- Amyloidose
- DCM (Dilatative Kardiomyopathie)
- Sportlerherz
- Angeborener Herzfehler mit Volumenüberlastung
z.B. Ebstein Anomalie, Vorhofseptumdefekt, Links-Rechts-Shunt, Perikardagenesie - Angeborener Morbus Uhl (angeborener hypoplastischer rechter Ventrikel)
- Idiopathische rechtsventrikuläre Ausflusstrakt-Tachykardie (RVOT-VT)
- Chagas Krankheit (Infektion durch Parasit Trypanosoma cruzi nach Biss von Raubwanzen)
- Zustand nach Rechtsherzinfarkt
- Cor pulmonale
- Ionenkanalerkrankungen
Long-QT-Syndrom, Short-QT-Syndrom, Brugada-Syndrom, CPVT = katecholaminerge polymorphe ventrikuläre Tachykardie)
- CAVE
Es ist äußerst wichtig, eine ARVC von ihren Mimics und möglichen Differentialdiagnosen abzugrenzen, einerseits um familiäre Erkrankung zu bestätigen oder auszuschließen, andererseits um eine adäquate Therapie einzuleiten, die sich von Fall zu Fall deutlich unterscheiden kann.
- Fehlende Familienanamnese (oft erster Hinweis auf genetische Erkrankung)
- Fehlinterpretation des EKG
- Fehlinterpretation des MRT (Über- und Unterdiagnose)
- Fehlinterpretation der Genetik (z.B. VUS)
- Unterlassen einer genetischen Untersuchung (z.B. bei Myokarditis)
- Mimics (z.B. Sarkoidose, die eine ARVC imitiert)
- inkomplette diagnostische Aufarbeitung
- CAVE
Nehmen Sie unspezifische Herzsymptome junger, vermeintlich gesunder PatientInnen ernst. Eine Familienanamnese bietet oft den ersten Hinweis auf eine familiäre Erkrankung und sollte auch bei einer Myokarditis erhoben werden. Im Zweifelfall sollte ein MRT durchgeführt und ggf. ein Gentest veranlasst werden.

- DCM (dilatative Kardiomyopathie)
- HCM (hypertrophe Kardiomyopathie)
- RCM (restriktive Kardiomyopathie)
- LVNC (Linksventrikuläre Non-compaction Kardiomyopathie)
- Morbus Fabry (Speicherkrankheit)
- Brugada-Syndrom
- andere Ionenkanalerkrankungen
- CAVE
Overlaps erschweren gelegentlich die Diagnosestellung. Im Zweifelsfall kann eine umfassende genetische Diagnostik zur Aufklärung beitragen.
Overlap mit anderen Kardiomyopathien
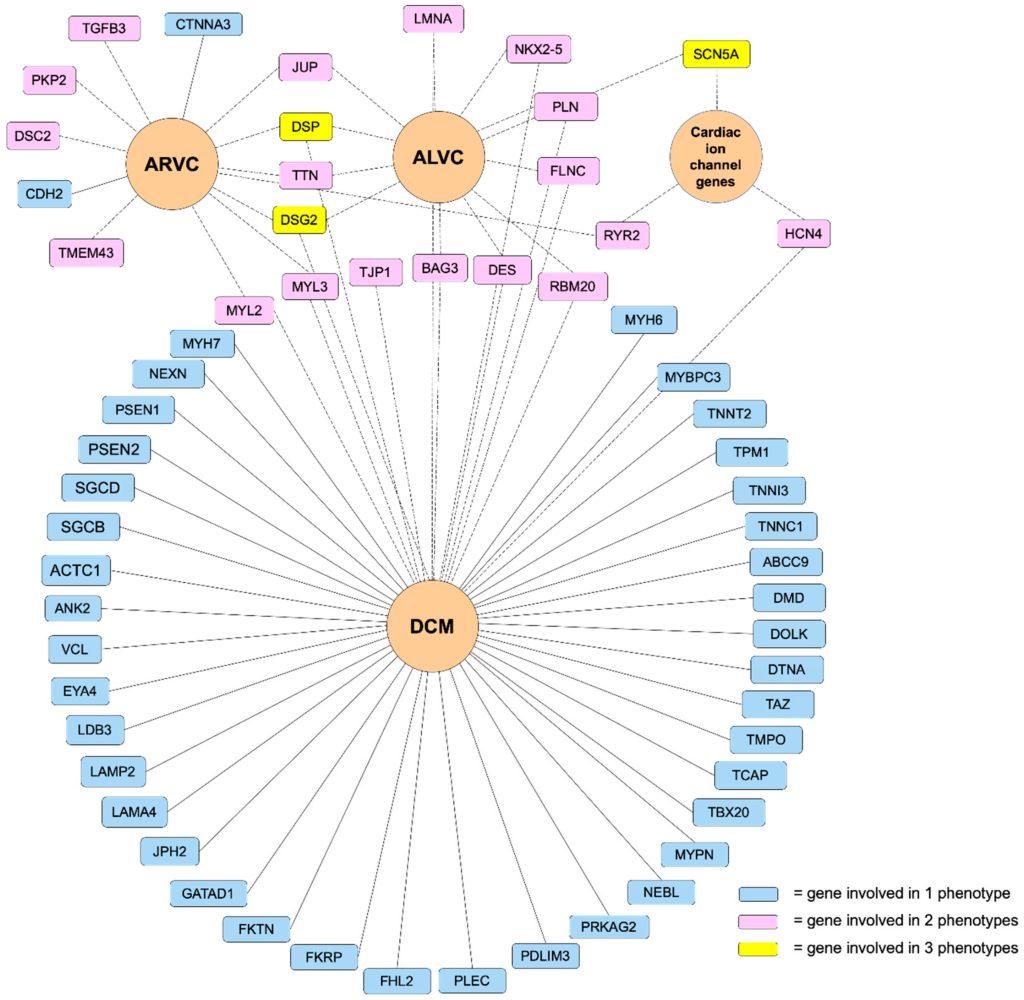
Overlap mit Ionenkanalerkrankungen und anderen Kardiomyopathien
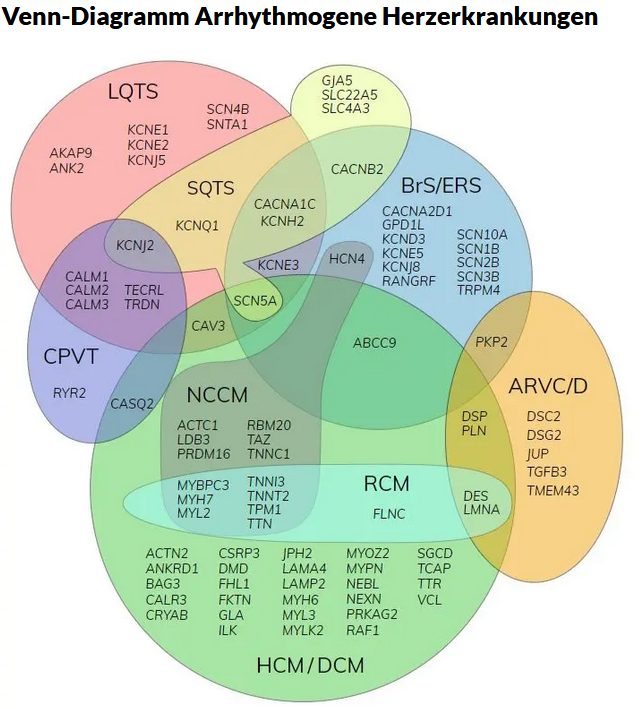
Wichtige Differentialdiagnosen
Mitunter schwierig ist die Abgrenzung einer ACM von einer Myokarditis, besonders, wenn es sich um die linksbetonten (ALVC) und beidseitigen Formen einer ACM (z.B. bei Trägern einer DSP-Genvariante) handelt, da bei diesen die typischen ARVC-Diagnosekriterien nicht immer greifen.
Besonders wenn es sich um eine rezidivierende Myokarditis mit fehlendem Virusnachweis handelt, sollte an eine ACM gedacht, eine ausführliche Eigen- und Familienanamnese erhoben und im Zweifelsfall ein Gentest durchgeführt werden.
Erfahrungen der ARVC-Selbsthilfe
Mehr als 10% unserer Mitglieder (und das sind nur diejenigen, von denen wir es wissen) wurden ursprünglich als Myokarditis diagnostiziert. Ob das jeweils eine Myokarditis war, die bei einem Anlageträger für ACM durch das Infektionsgeschehen den Ausbruch der Erkrankung getriggert, oder ob es sich um eine sogenannte "hot phase" einer ACM gehandelt hat, bleibt dabei unklar. Auf alle Fälle hätte bei vielen dieser Mitglieder bereits eine ausführliche Familien- und Eigenanamnese einen Hinweis auf die Diagnose erbracht. Durch einen Gentest wurde dann in der Mehrzahl der Fälle die endgültige Diagnose ARVC/ACM/ALVC gestellt.
Tatsächlich finden sich in unter unseren Mitgliedern aber auch solche, die eine bioptisch gesicherte eosinophile Myokarditis oder eine Myokarditis MIT positivem Virusnachweis hatten. Daher: lieber ein Gentest zu viel als einer zu wenig.
Eine Abgrenzung zur DCM ist insbesondere bei den linksbetonten Formen der ACM nicht immer einfach. Eine exakte Diagnosestellung ist insofern wichtig, da bei DCM meist kein so hohes Risiko für Herzrhythmusstörungen besteht, was sehr wichtig für die individuelle Risikoeinschätzung ist.
- EKG:
Die für ACM im EKG typische Niedervoltage (niedrige Amplitude des QRS-Komplexes) in den Extremitätenableitungen, negative T-Wellen in den inferolateralen Ableitungen und ausgeprägte ventrikuläre Arrhythmien sind bei der DCM seltener zu finden. - MRT:
Im MRT sieht man neben einer verringerten LVEF bei beiden Erkrankungen ein Late-Gadolinium-Enhancement (LGE), das Ausdruck des bindegewebigen Umbaus des Herzmuskels ist. Das LGE ist allerdings bei ACM deutlicher ausgeprägt und meist subepikardial lokalisiert. Ein linksventrikuläres LGE ≥20% tritt praktisch nur bei ACM auf. - Gentest:
Im Zweifelsfall kann ein Gentest mit einem Kardiomyopathie-Panel und dem Auffinden einer ACM- oder DCM-typischen Genvariante zur Abklärung beitragen.
- Familienanamnese
Leer
- 12-Kanal-EKG
normal - Echo/MRT
Keine strukturellen Auffälligkeiten
- EPU
keine strukturellen Auffälligkeiten im Mapping
monomorphe VT
hochdosierte Isoproterenol-Gabe unauff.
keine VTs bei programmierter ventrikulärer Stimulation induzierbar - Prognose
benigne
Ein vergrößertes Herz kann grundsätzlich auch eine physiologische Adaption an das Training sein. Die Vergrößerung des rechten Ventrikels, EKG-Besonderheiten und Arrhythmien können die hämodynamische Belastung durch den Sport widerspiegeln.
- Familienanamnese
Leer - 12-Kanal-EKG
häufige VES
invertierte T-Wellen in präkordialen Ableitungen - Echo/MRT
Keine offensichtlichen strukturellen Auffälligkeiten
↔ bei ARVC: globale systolische Dysfunktion des RV und/oder regionale Wandbewegungsstörungen im RV wie Aneurysmen - Prognose
benigne
Mehr erfahren
Task-Force-Diagnosekriterien ARVC
Fachartikel Diagnose
Vortragsfolien „Rare Diseases: Underdiagnosed and undertreated?“, Prof. Dr. med. Thomas Wichter (2018)
YouTube-Kanal Ärztefortbildung zu ARVC/ACM
Video Ärztefortbildung „ACM/ARVC – Diagnosekriterien up-to-date“ PD Dr. med. Sebastian Clauß (2023)
Video Ärztefortbildung „Kardio-Bildgebung bei ACM – MRT“, Prof. Dr. med. Stefan Kääb/Dr. med. Felix Escher (2023)
Video des Vortrags „Diagnose der ARVC – Schwerpunkt MRT“, Prof. Dr. med. Jeanette Schulz-Menger (2023)
Pocket-Leitlinie: Kardiomyopathien - Leitlinien für das Management von Kardiomyopathien (Version 2023)
Meder B, Eckardt L, Falk V et al. Deutsche Gesellschaft für Kardiologie – Herz- und Kreislaufforschung e.V. (2023) - ESC Pocket Guidelines, Börm Bruckmeier Verlag GmbH
https://leitlinien.dgk.org/2024/pocket-leitlinien-kardiomyopathien-version-2023/
2023 ESC Guidelines for the management of cardiomyopathies
Arbelo E, Protonotarios A, Gimeno JR et al. ESC Scientific Document Group, Eur Heart J. 2023 Aug 25:ehad194
https://doi.org/10.1093/eurheartj/ehad194
Video der Ärztefortbildung „ACM/ARVC - Diagnosekriterien up-to-date“
PD Dr. med. Sebastian Clauß, Klinikum der LMU München
Ärztefortbildung des Munich Consortiums des European Reference Networks ERN GUARD-Heart „Arrhythmogene Kardiomyopathien (ACM)“ in Zusammenarbeit mit dem ARVC-Selbsthilfe e.V., St. Vinzenz Haus München, 16.06.2023
https://www.youtube.com/watch?v=6eTe6AZT9w8
Video der Ärztefortbildung „Kardio-Bildgebung bei ACM - MRT“
Prof. Dr. med. Stefan Kääb/Dr. med. Felix Escher, Klinikum der LMU München
Ärztefortbildung des Munich Consortiums des European Reference Networks ERN GUARD-Heart „Arrhythmogene Kardiomyopathien (ACM)“ in Zusammenarbeit mit dem ARVC-Selbsthilfe e.V., St. Vinzenz Haus München, 16.06.2023
https://www.youtube.com/watch?v=_lwni7D-8tU
Video der Ärztefortbildung „ACM und Arrhythmien – Risikostratifizierte Therapieoptionen“
PD Dr. med. Moritz Sinner, Klinikum der LMU München
Ärztefortbildung des Munich Consortiums des European Reference Networks ERN GUARD-Heart „Arrhythmogene Kardiomyopathien (ACM)“ in Zusammenarbeit mit dem ARVC-Selbsthilfe e.V., St. Vinzenz Haus München, 16.06.2023
https://www.youtube.com/watch?v=PntnAeaeBkY
Video der Ärztefortbildung „ACM: Kausale Gene, Umgang mit Varianten unklarer Signifikanz (VUS) und genetische Diagnostik bei Kindern“
PD Dr. med. Dominik Westphal, Klinikum rechts der Isar der TU München
Ärztefortbildung des Munich Consortiums des European Reference Networks ERN GUARD-Heart „Arrhythmogene Kardiomyopathien (ACM)“ in Zusammenarbeit mit dem ARVC-Selbsthilfe e.V., St. Vinzenz Haus München, 16.06.2023
https://www.youtube.com/watch?v=XprLLxfLHso
Pocket-Leitlinie: Ventrikuläre Arrhythmien und Prävention des plötzlichen Herztodes (Version 2022)
Eckardt L, Bosch R, Falk V, et al. Deutsche Gesellschaft für Kardiologie – Herz- und Kreislaufforschung e.V. (2023) - ESC Pocket Guidelines, Börm Bruckmeier Verlag GmbH
https://leitlinien.dgk.org/2023/pocket-leitlinie-ventrikulaere-arrhythmien-und-praevention-des-ploetzlichen-herztodes-version-2022/
2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death
Zeppenfeld K, Tfelt-Hansen J, de Riva M et al. Eur Heart J. 2022 Oct 21;43(40):3997-4126
https://doi.org/10.1093/eurheartj/ehac262
Arrhythmogenic left ventricular cardiomyopathy
Corrado D, Basso C; Heart. 2021 Jul 13:heartjnl-2020-316944
https://oi.org/10.1136/heartjnl-2020-316944
‘Hot phase’ clinical presentation in arrhythmogenic cardiomyopathy
Bariani R, Cipriani A, Rizzo S et al. EP Europace, Volume 23, Issue 6, June 2021, Pages 907–917
https://doi.org/10.1093/europace/euaa343
Arrhythmogenic Right Ventricular Cardiomyopathy Presenting as Clinical Myocarditis in Women
Scheel PJ3rd, Murray B, Tichnell C et al. Am J Cardiol. 2021 Jan 15:S0002-9149(21)00048-5
https://doi.org/10.1016/j.amjcard.2020.12.090
Desmoplakin Cardiomyopathy, a Fibrotic and Inflammatory Form of Cardiomyopathy Distinct From Typical Dilated or Arrhythmogenic Right Ventricular Cardiomyopathy
Smith ED, Lakdawala NK, Papoutsidakis N et al. Circulation. 2020;141(23):1872-1884
https://doi.org/10.1161/CIRCULATIONAHA.119.044934
Arrhythmogenic Right Ventricular Cardiomyopathy: Evaluation of the Current Diagnostic Criteria and Differential Diagnosis
Corrado D, van Tintelen PJ, McKenna WJ et al. Eur Heart J. 2020 Apr 7;41(14):1414-1429
https://doi.org/10.1093/eurheartj/ehz669
Arrhythmogenic Right Ventricular Cardiomyopathy: Characterization of Left Ventricular Phenotype and Differential Diagnosis With Dilated Cardiomyopathy
Cipriani A, Bauce B, De Lazzari M et al. J Am Heart Assoc. 2020 Mar 3;9(5):e014628
https://doi.org/10.1161/JAHA.119.014628
Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria
Corrado D, Perazzolo Marra M, Zorzi A. et al. Int J Cardiol. 2020;S0167-5273(20)33293-9
https://doi.org/10.1016/j.ijcard.2020.06.005
2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy
Towbin JA, McKenna WJ, Abrams DJ et al. Heart Rhythm Volume 16, ISSUE 11, e301-e372, November 01, 2019
https://doi.org/10.1016/j.hrthm.2019.05.007
Vortrag „Rare Diseases: Underdiagnosed and undertreated?“
Prof. Dr. med. Thomas Wichter, ehemaliger Chefarzt Niels-Stensen-Kliniken, Marienhospital Osnabrück, Klinik für Innere Medizin und Kardiologie
Jahrestagung der Deutschen Gesellschaft für Kardiologie - Herz- und Kreislaufforschung e.V. (DGK), Mannheim, 06.04.2018
Vortragsfolien
Arrhythmogenic Right Ventricular Cardiomyopathy: An Update on Pathophysiology, Genetics, Diagnosis, and Risk Stratification
Paul M, Wichter T, Fabritz L et al. Herzschrittmacherther Elektrophysiol. 2012 Sep;23(3):186-95
https://doi.org/10.1007/s00399-012-0233-7
Die arrhythmogene rechtsventrikuläre Dysplasie/Kardiomyopathie
Saguner A, Brunckhorst C, Duru F. Cardiovascular Medicine 2011; 14 (11): 303-314
https://cardiovascmed.ch/article/doi/cvm.2011.01623
Diagnosis of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia - Proposed Modification of the Task Force Criteria
Marcus FI, McKenna WJ, Sherrill D et al. Circulation. 2010;121:1533–1541
https://doi.org/10.1161/CIRCULATIONAHA.108.840827
Letzte Aktualisierung: 17.10.2024